Welcome to the b2slab
Bioinformatics and Biomedical Signals Laboratory
This is a research group member of the Institut de Recerca i Innovació en Salut (IRIS) of the Universitat Politècnica de Catalunya (UPC) in Barcelona, Spain. Former member of the Research Center for Biomedical Engineering (CREB)
Our main three research lines are :
- Analysis of large datasets from primary and non-primary care related datasets.
- Methods in Bioinformatics, specially in metabolomics
- Biomedical Engineering
The Bioinformatics and Biomedical Signals Laboratory (B2SLab) at the Universitat Politècnica de Catalunya (UPC) focuses on the intersection of bioinformatics and biomedical signal processing. Our research encompasses areas such as computational modeling, machine learning, and data analysis applied to biological and medical data.
Check:
Use cases
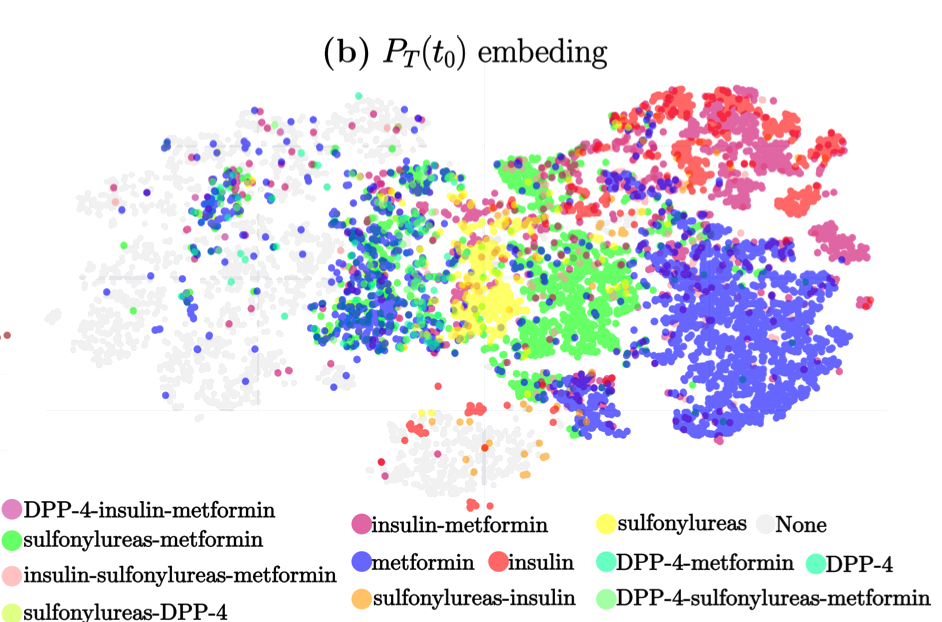
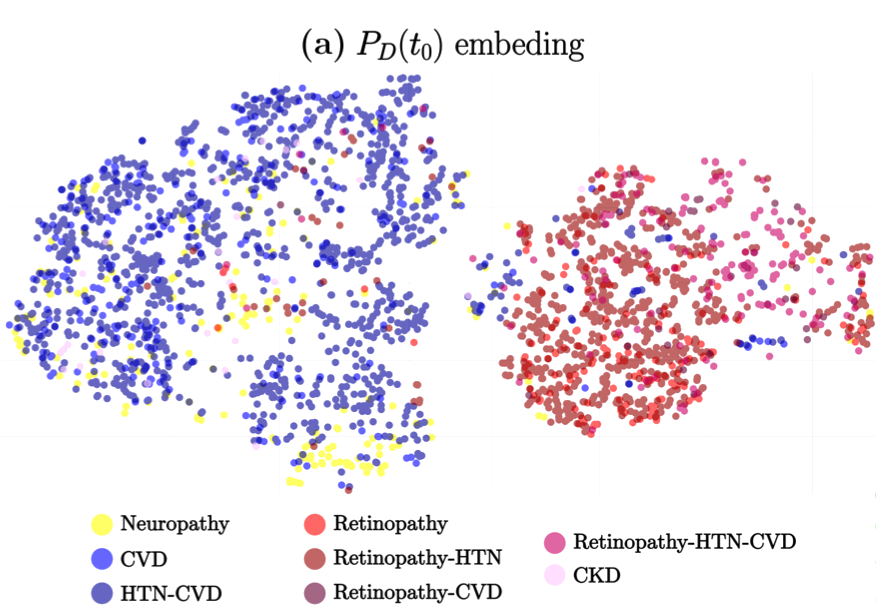
The study titled “A Deep Attention-Based Encoder for the Prediction of Type 2 Diabetes Longitudinal Outcomes from Routinely Collected Health Care Data” introduces DARE (Diabetic Attention with Relative position Representation Encoder), a transformer-based model designed to analyze longitudinal and heterogeneous data of Type 2 Diabetes Mellitus (T2DM) patients. Leveraging data from over 200,000 diabetic individuals in the SIDIAP primary healthcare database, which includes diagnostic codes, medication records, and various clinical measurements, DARE underwent an unsupervised pre-training phase followed by fine-tuning for three specific clinical prediction tasks:
Predicting the occurrence of comorbidities. Assessing the achievement of target glycemic control (defined as glycated hemoglobin < 7%). Forecasting changes in glucose-lowering treatments. In cross-validation, DARE demonstrated superior performance compared to baseline models, achieving area under the curve (AUC) scores of 0.88 for comorbidity prediction, 0.91 for treatment prediction, and 0.82 for HbA1c target prediction. These findings suggest that attention-based encoders like DARE can effectively model complex relationships in longitudinal T2DM data, potentially aiding clinicians in personalized disease management strategies.
The study titled “mWISE: An Algorithm for Context-Based Annotation of Liquid Chromatography–Mass Spectrometry Features through Diffusion in Graphs” introduces mWISE, an R package designed to enhance the annotation of LC–MS data in untargeted metabolomics. Traditional annotation methods often face challenges due to the complexity and volume of data generated in metabolomics studies. mWISE addresses these challenges by employing a context-based approach that utilizes diffusion in graphs to improve the accuracy and efficiency of feature annotation. This method allows for a more comprehensive understanding of the metabolome by effectively integrating various data sources and contextual information.

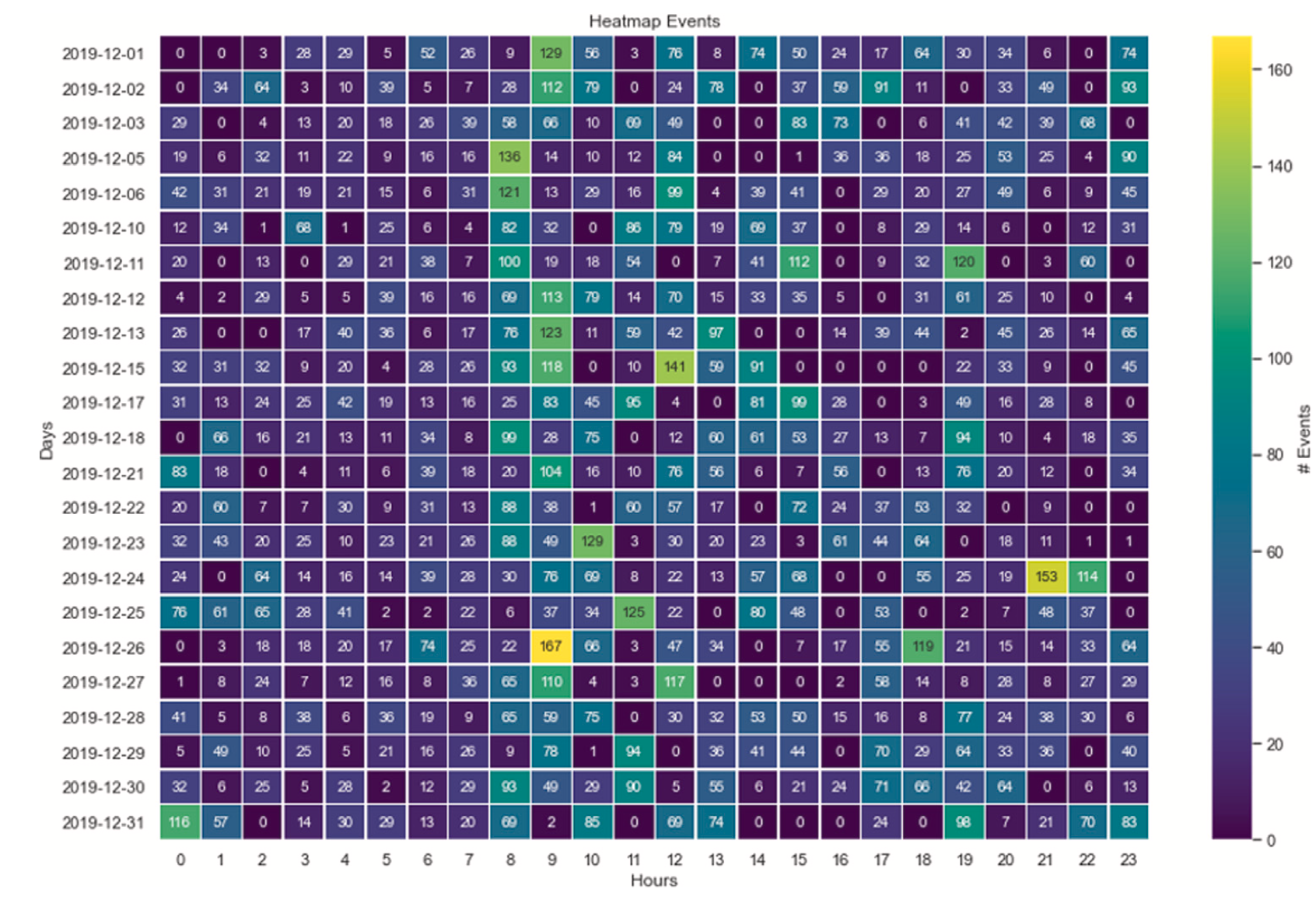
This study explores the feasibility of using gas sensor arrays for unobtrusive home monitoring of elderly individuals living alone. Unlike traditional monitoring systems, such as cameras or motion sensors, gas sensors provide a non-invasive and privacy-preserving alternative by detecting changes in air composition associated with human activities. The researchers developed a wireless sensor unit incorporating metal oxide gas sensors, carbon dioxide sensors, carbon monoxide sensors, and temperature-humidity sensors, which was deployed in an elderly person’s home for three months. The system continuously monitored the indoor air environment and used Principal Component Analysis (PCA) and statistical anomaly detection to extract activity patterns while compensating for environmental drift. The findings confirmed that gas sensors effectively captured daily routines and detected deviations—such as a Christmas Eve family gathering—demonstrating their ability to monitor Activities of Daily Living (ADLs) and recognize anomalies that could indicate emergencies or changes in well-being.
Compared to motion sensors, the gas sensor system offered broader coverage without blind spots, as changes in air composition spread throughout the living space. The correlation between gas sensor data and motion sensor recordings validated its effectiveness. However, some limitations were noted, including delayed event detection due to air dispersion and the need for multiple sensor nodes for room-specific monitoring. The study suggests that integrating gas sensors into IoT-based smart home systems could enhance elderly care by providing real-time activity tracking, anomaly detection, and early warning signals to caregivers and family members. Future research should focus on improving spatial resolution, refining machine learning models for predictive analysis, and combining gas sensors with other smart home technologies to create a more robust, adaptive, and personalized home monitoring solution.