
Time-Series & Sequence Modeling with RNNs
Advanced Topics in Machine Learning for Bioinformatics and Biomedical Engineering
Dr. Alexandre Perera Lluna 
April 20, 2026
Why sequence models?
Biological sequences are everywhere:
- DNA:
ATCGATCGTA… - Proteins:
MKTAYIAKQR… - ECG: \(x_1, x_2, \ldots, x_T\)
- EEG signals over time
- Gene expression time-courses

Sequential biological data: four modalities
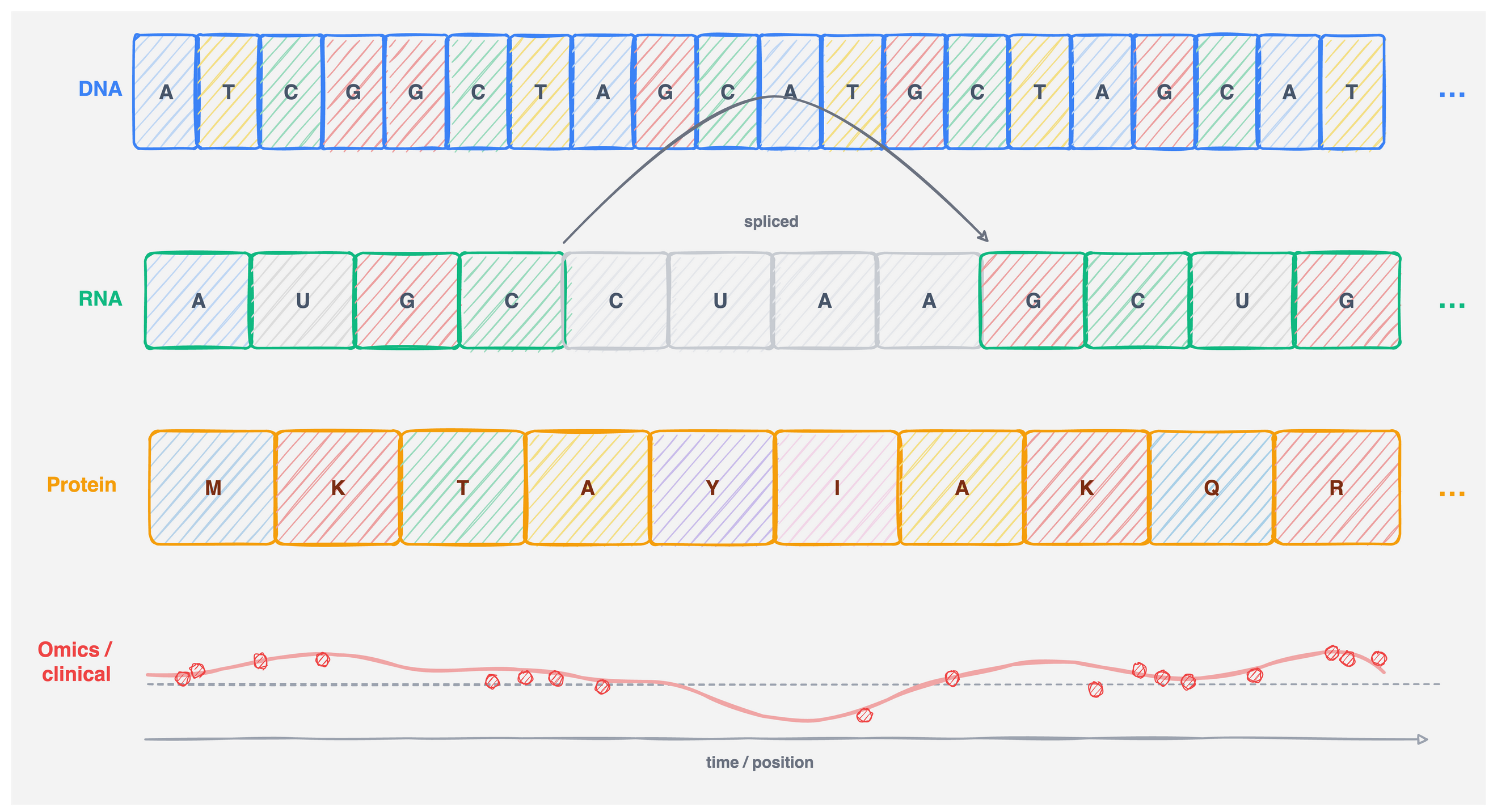
Figure 2: Four sequential modalities encountered in bioinformatics. Each row represents one data type; the horizontal axis is always position or time.
MLP limitation 1: fixed input size
Fixed Input Size
An MLP with input layer of width \(d\) requires every input to have exactly \(d\) features. There is no mechanism to handle variable-length sequences natively.
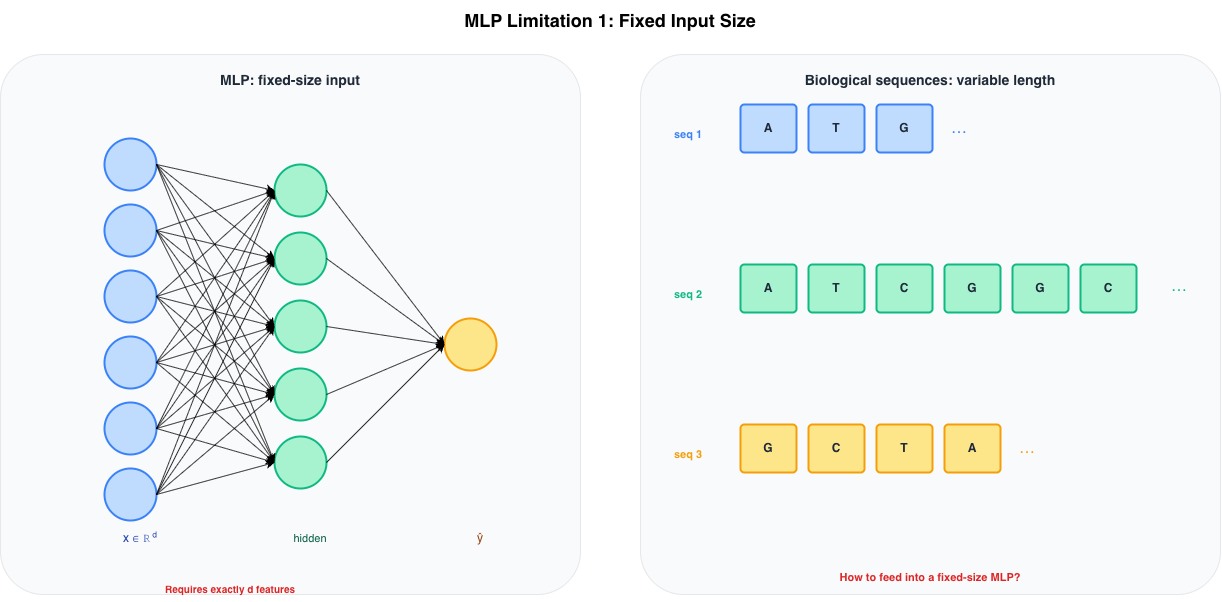
Figure 3: Left: an MLP requires exactly d input features. Right: biological sequences come in variable lengths — three sequences of lengths 3, 8 and 4 cannot share the same fixed-width input layer.
Fixed Input Size
The two engineering workarounds both lose information:
- Truncation: discard positions beyond a fixed cutoff – relevant context is dropped
- Padding + flattening: pad short sequences to length \(T_{\max}\) – the model must waste capacity learning to ignore padding
Figure 4: MLP fixed-size input vs variable-length sequences.
MLP limitation 2: no memory across positions
Even if we fix the length problem – e.g., by applying an MLP independently at each position)– the MLP has no way to pass information from one position to the next.
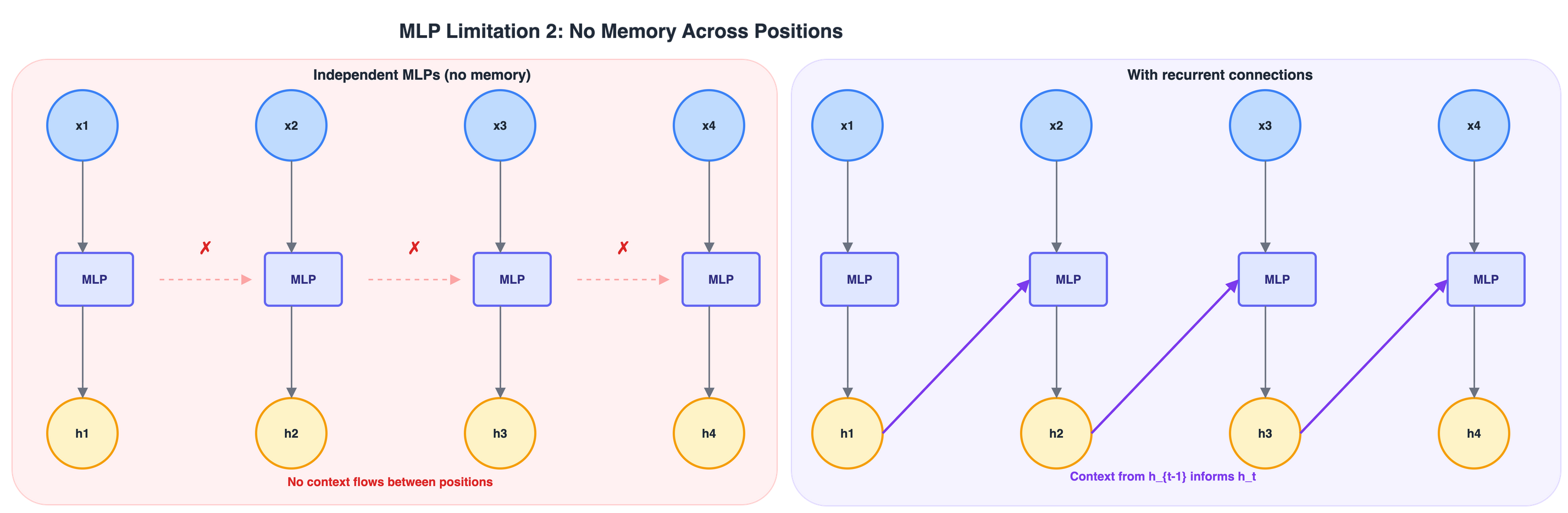
Figure 5: Left: independent MLPs applied at each position share no information (\(\times\)). Right: recurrent connections allow context from \(h_{t-1}\) to influence \(h_t\) (purple arrows).
MLP limitation 2: consequences for biology
For example, an MLP applied position-by-position cannot:
- Detect a motif that spans multiple positions without seeing all positions simultaneously (defeats variable-length handling)
- Track codon reading frame – the interpretation of position \(t\) depends on \(t \bmod 3\), which requires knowing where the sequence started
- Model splice site context – both the donor and acceptor site must be jointly considered, though they are tens to thousands of nucleotides apart
- Capture promoter–TSS distance – a regulatory element’s effect depends on its distance from the transcription start, not on its local identity alone

The sliding window
Before recurrent networks, the standard approach was a sliding window: extract a fixed-width context of \(k\) positions around each target position and apply an MLP to that window.
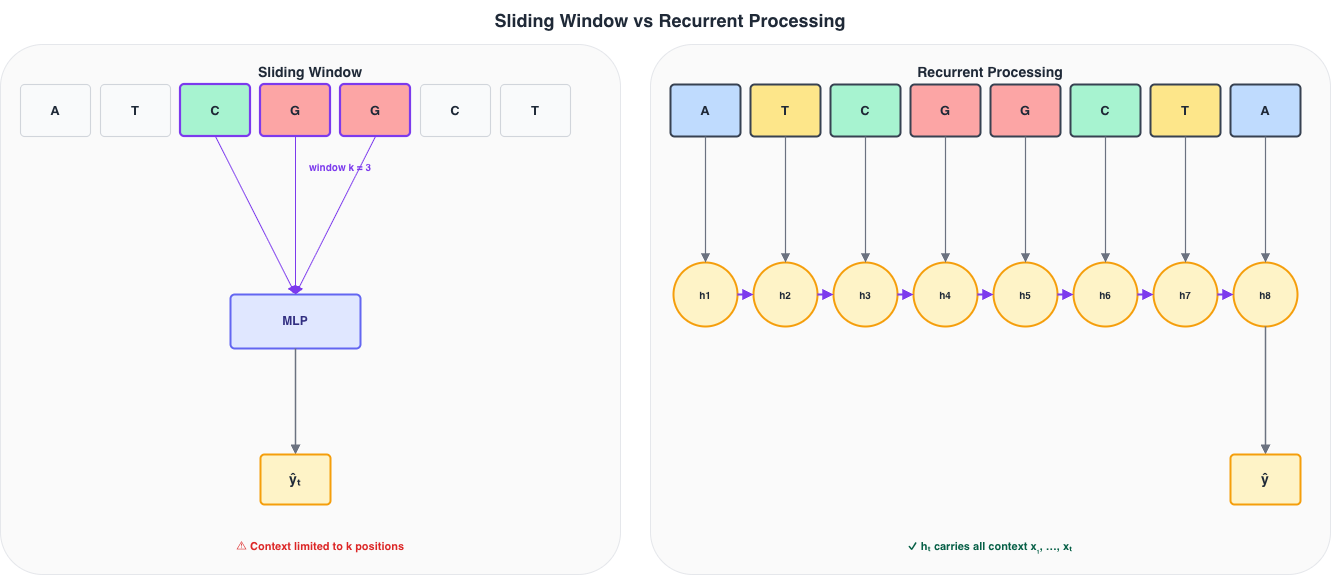
Figure 6: Left: a sliding window of size k=3 extracts a local context and applies an MLP. Right: a recurrent model accumulates all past context in h_t without a fixed horizon.
Sequence mapping
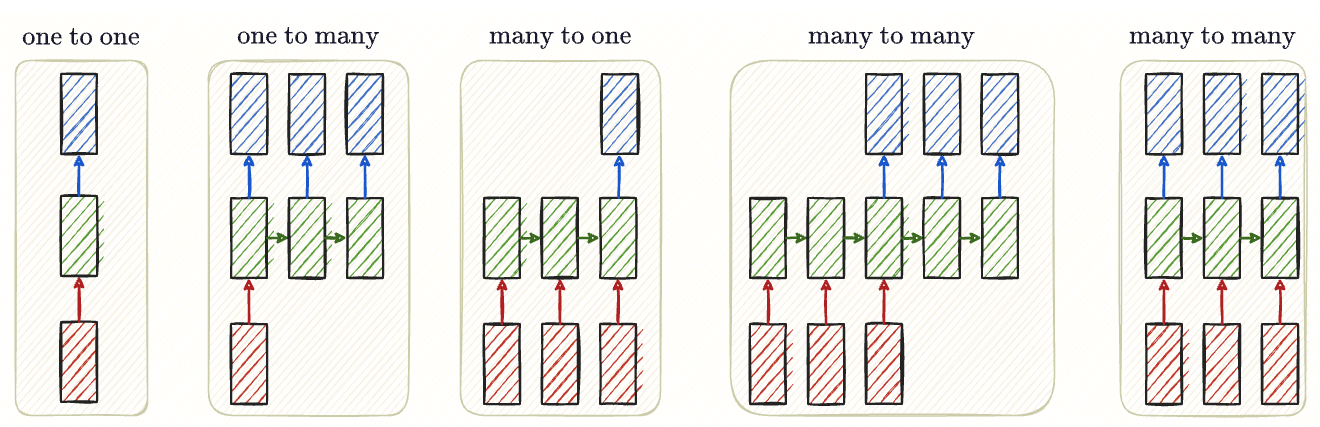
Generally, we will find these sequence mapping variants
Sequence task settings
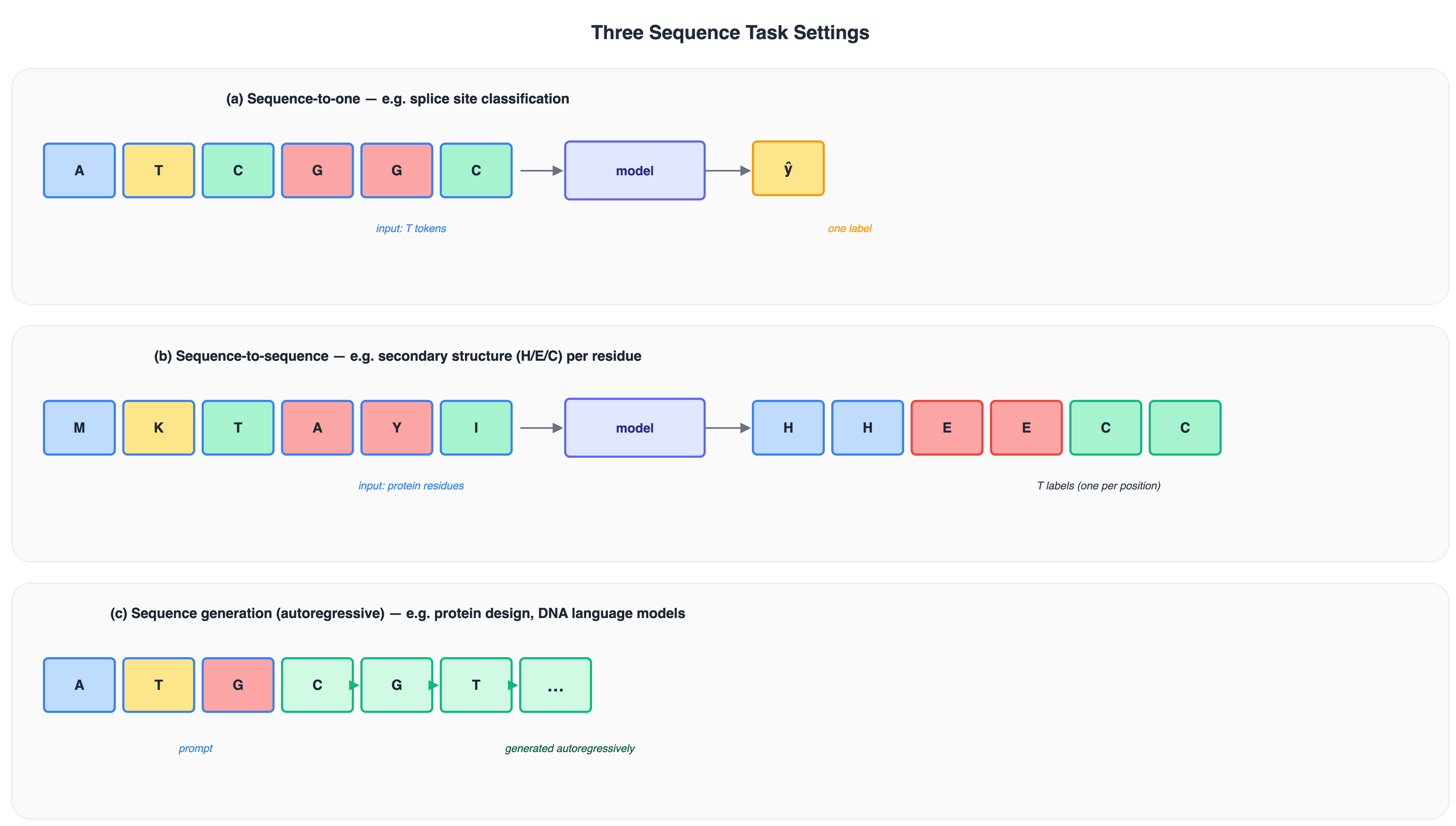
Figure 7: Three standard sequence task settings. (a) One label for the whole sequence. (b) One label per position. (c) Tokens generated one at a time, each conditioned on all previous tokens.
Parameter sharing across time
The function \(f\) uses the same parameters at every time step. For the simplest recurrent model:
\[ h_t = \phi\!\left(W_x\, x_t + W_h\, h_{t-1} + b\right) \]
where \(W_x \in \mathbb{R}^{d_h \times d_{\text{in}}}\), \(W_h \in \mathbb{R}^{d_h \times d_h}\), and \(b \in \mathbb{R}^{d_h}\) are shared across all \(T\) steps.
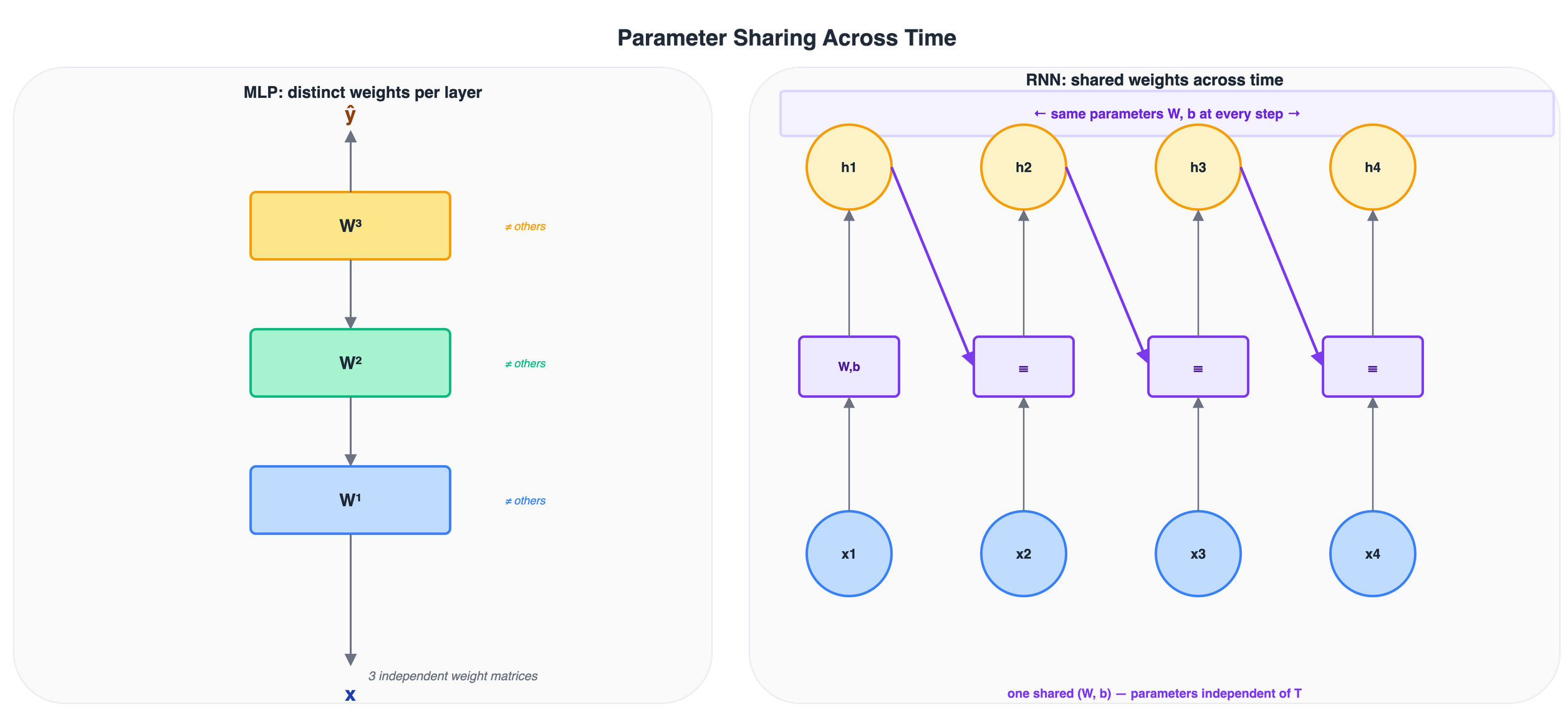
Figure 8: Left: an MLP uses distinct weight matrices at each layer. Right: a recurrent model reuses the same (W, b) at every time step, keeping the parameter count independent of sequence length T.
The RNN unit
At each time step \(t\), the unit receives two inputs:
- \(x_t \in \mathbb{R}^{d_{\text{in}}}\) — the current observation
- \(h_{t-1} \in \mathbb{R}^{d_h}\) — the hidden state carried from the previous step
It produces a new hidden state:
\[ h_t = \tanh(W_h h_{t-1} + W_x x_t + b) \]
- \(W_h \in \mathbb{R}^{d_h \times d_h}\) mixes the recurrent memory
- \(W_x \in \mathbb{R}^{d_h \times d_{\text{in}}}\) projects the current input
- \(\tanh\) keeps values in \((-1, 1)\), preventing unbounded growth
- \(h_t\) is both the output at step \(t\) and the input to step \(t+1\)
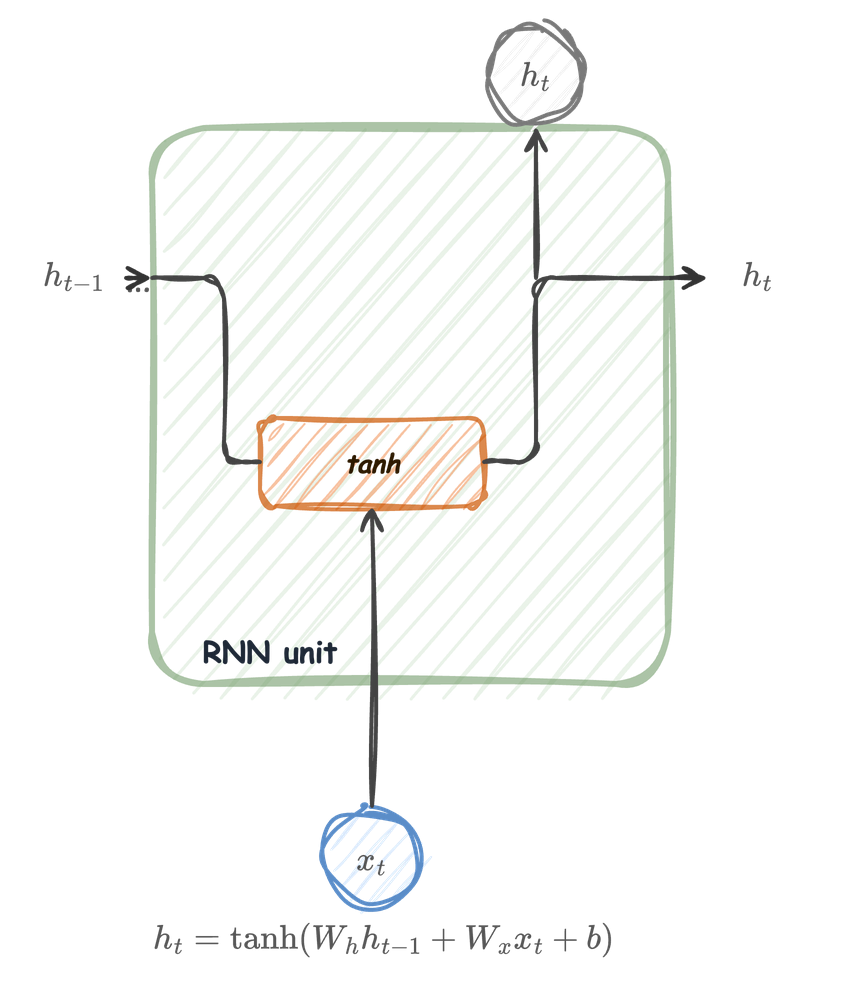
The RNN unit
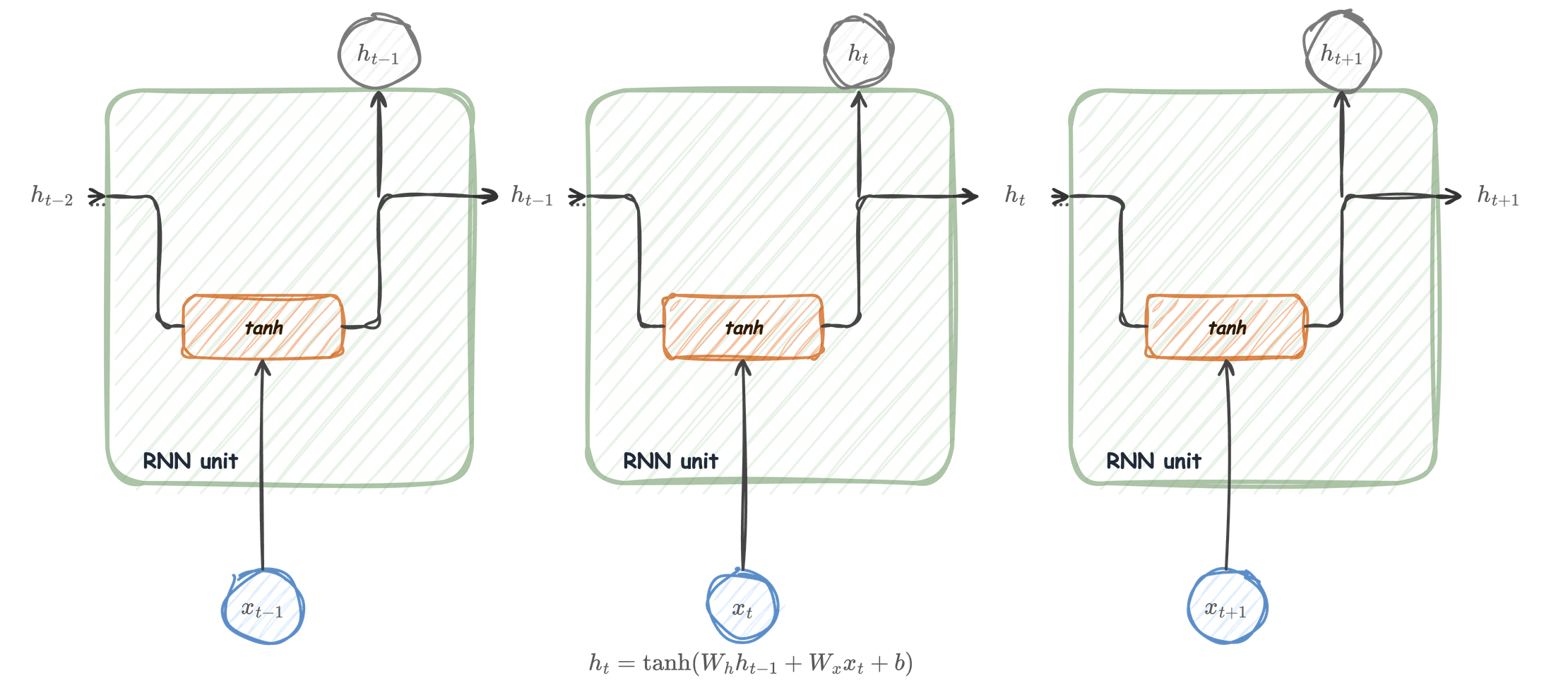
Unfolded
A numerical example
Effect of clipping on the gradient landscape

The LSTM cell at a glance
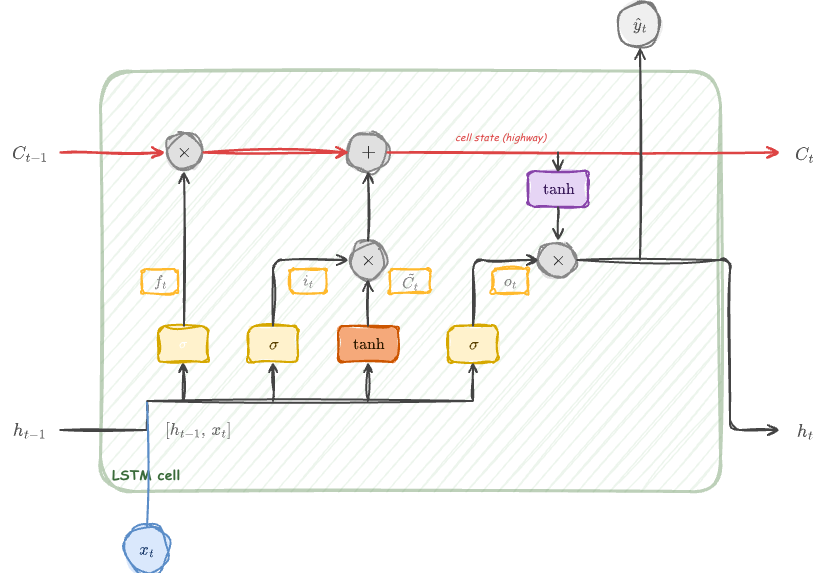
LSTM cell: the cell state \(C_t\) flows along the top as a highway. Four components — forget gate (\(f_t\)), input gate (\(i_t\)), candidate (\(\tilde{C}_t\)), and output gate (\(o_t\)) — regulate information flow.
Forget gate

The forget gate reads \([h_{t-1}, x_t]\) and produces \(f_t\) through a sigmoid. It then multiplies \(C_{t-1}\) element-wise, controlling which memory dimensions survive. Forget gate biases are often initialized to 1.0 so the network starts by remembering, not forgetting.
Forget gate
\[ f_t = \sigma\!\bigl(W_f\, [h_{t-1},\, x_t] + b_f\bigr) \]
Dimensions:
- \([h_{t-1}, x_t] \in \mathbb{R}^{d_h + d_{\text{in}}}\)
- \(W_f \in \mathbb{R}^{d_h \times (d_h + d_{\text{in}})}\), \(b_f \in \mathbb{R}^{d_h}\)
- \(f_t \in (0, 1)^{d_h}\): one val. per dim of \(C_{t-1}\)
Per-dimension behaviour:
- \(f_t^{(j)} \approx 1\): dim \(j\) preserved
- \(f_t^{(j)} \approx 0\): dim \(j\) erased
Input gate
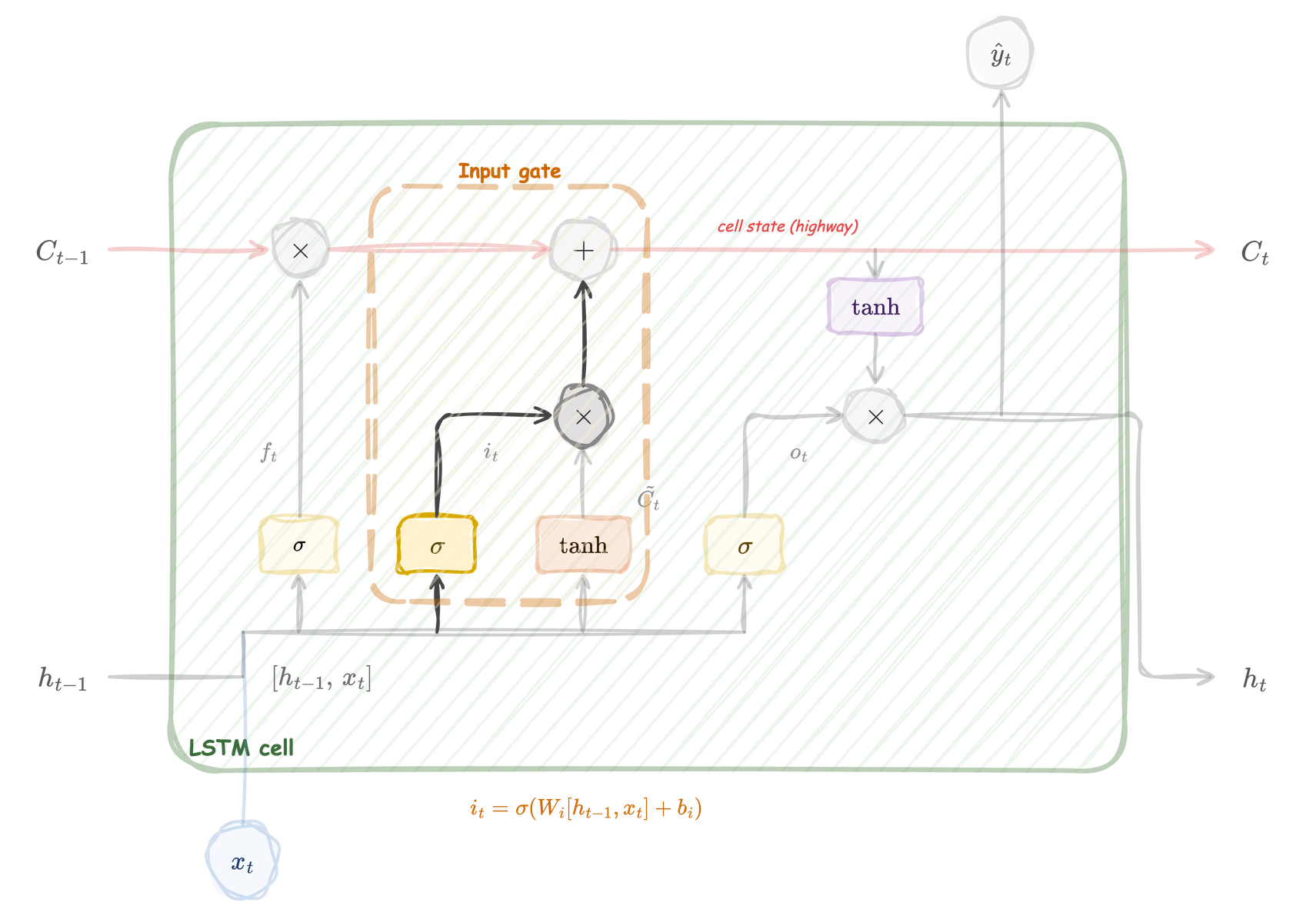
The input gate reads \([h_{t-1}, x_t]\) through a sigmoid and produces \(i_t\). It controls how much of the new candidate to write into the cell state. The input gate is the volume knob; the candidate (next) provides the content.
Input gate
\[ i_t = \sigma\!\bigl(W_i\, [h_{t-1},\, x_t] + b_i\bigr) \]
Dimensions:
- \(W_i \in \mathbb{R}^{d_h \times (d_h + d_{\text{in}})}\), \(b_i \in \mathbb{R}^{d_h}\)
- \(i_t \in (0,1)^{d_h}\): per-dimension write strength
Separation of roles:
- Input gate decides how much to write (control)
- Candidate decides what to write (content)
Candidate value

The candidate computes \(\tilde{C}_t\) through a \(\tanh\) activation. Unlike the three gates, it proposes content rather than control. Combined with the input gate as \(i_t \odot \tilde{C}_t\), it determines what new information enters the cell state.
Candidate value
\[ \tilde{C}_t = \tanh\!\bigl(W_C\, [h_{t-1},\, x_t] + b_C\bigr) \]
Dimensions:
- \(W_C \in \mathbb{R}^{d_h \times (d_h + d_{\text{in}})}\), \(b_C \in \mathbb{R}^{d_h}\)
- \(\tilde{C}_t \in (-1, 1)^{d_h}\): proposed content
Key properties:
- Uses \(\tanh\) (+,-)
- Only non-gate component
- Gated contribution: \(i_t \odot \tilde{C}_t\)
Output gate

The output gate selects which dimensions of the squashed cell state \(\tanh(C_t)\) are exposed as \(h_t\). The cell can store information internally that it does not yet reveal — like a notebook where you choose which page to show.
Output gate
\[ o_t = \sigma\!\bigl(W_o\, [h_{t-1},\, x_t] + b_o\bigr), \qquad h_t = o_t \odot \tanh(C_t) \]
Dimensions:
- \(W_o \in \mathbb{R}^{d_h \times (d_h + d_{\text{in}})}\), \(b_o \in \mathbb{R}^{d_h}\)
- \(o_t \in (0, 1)^{d_h}\)
Two-step output:
- \(\tanh(C_t)\) \((-1, 1)\)
- \(o_t\) selects dims to reveal
- \(h_t\) feeds preds and next step
\(C_t\) can store values outside \((-1,1)\) because only the output view \(h_t\) is squashed. This gives \(C_t\) more dynamic range.
Putting it all together

Complete LSTM cell with all gates active. The red arrow shows the cell state highway; the four blocks at the bottom compute the gates and candidate from \([h_{t-1}, x_t]\).
Putting it all together
All four equations for one time step:
\[\begin{eqnarray} f_t &=& \sigma\bigl(W_f [h_{t-1}, x_t] + b_f\bigr) \\ i_t &= & \sigma\bigl(W_i [h_{t-1}, x_t] + b_i\bigr)\\ \tilde{C}_t &= & \tanh\bigl(W_C [h_{t-1}, x_t] + b_C\bigr)\\ C_t &= &f_t \odot C_{t-1} + i_t \odot \tilde{C}_t\\ o_t &= & \sigma\bigl(W_o [h_{t-1}, x_t] + b_o\bigr)\\ h_t &= & o_t \odot \tanh(C_t) \end{eqnarray}\]
Inputs: \(h_{t-1}, C_{t-1}, x_t\). Outputs: \(h_t, C_t\).
Gradient flow: a visual comparison

The GRU cell at a glance
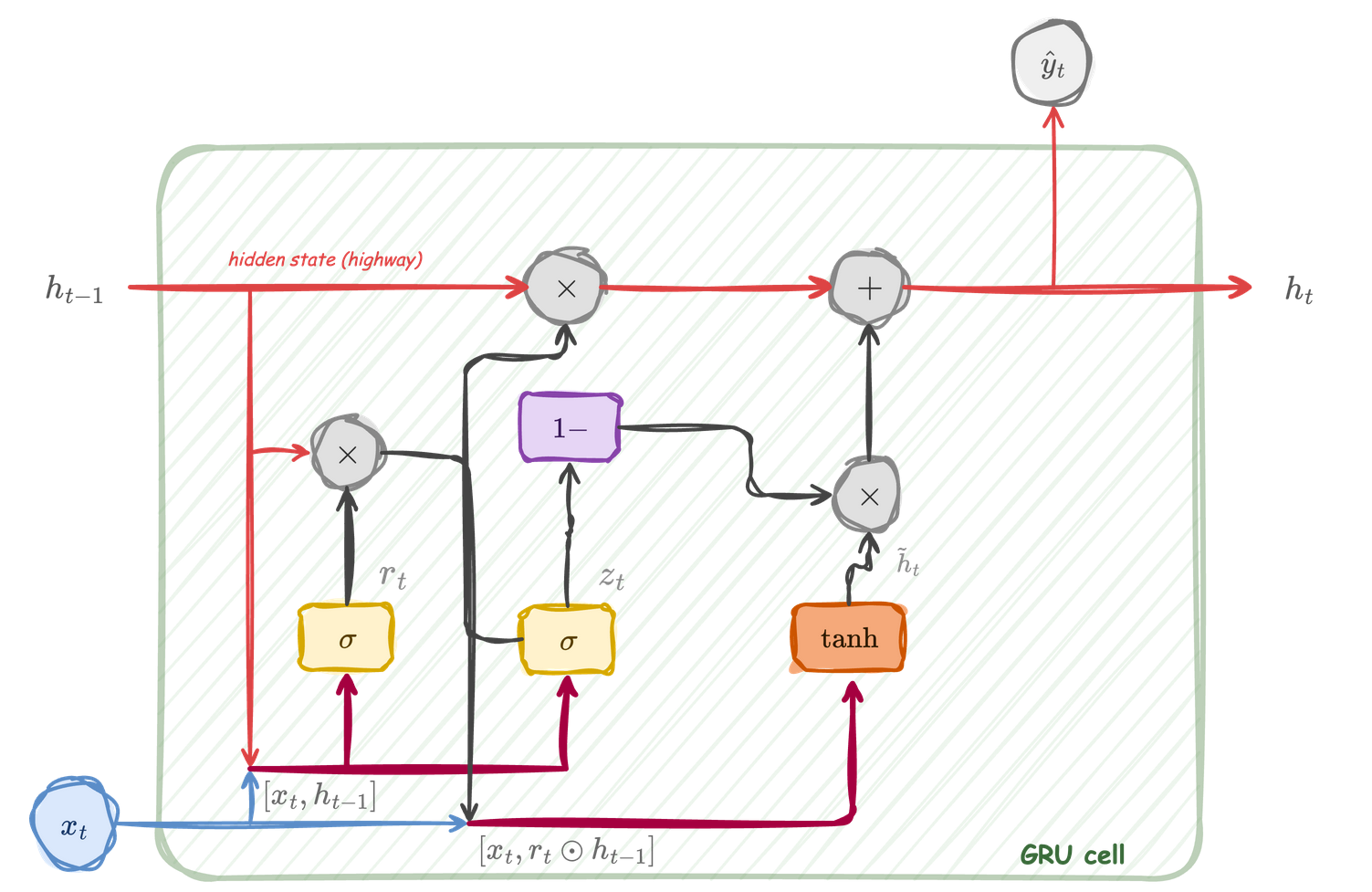
GRU cell: the hidden state \(h_t\) is updated through a convex combination controlled by the update gate \(z_t\). The reset gate \(r_t\) modulates how much of \(h_{t-1}\) influences the candidate \(\tilde{h}_t\).
Reset gate

The reset gate reads \([h_{t-1}, x_t]\) and produces \(r_t\) through a sigmoid. It controls how much of the previous hidden state flows into the candidate computation. When \(r_t \approx 0\), the candidate ignores the past and behaves like a plain feed-forward layer.
Reset gate
\[ r_t = \sigma\bigl(W_r\, [h_{t-1},\, x_t] + b_r\bigr) \]
Dimensions:
- \([h_{t-1}, x_t] \in \mathbb{R}^{d_h + d_{\text{in}}}\)
- \(W_r \in \mathbb{R}^{d_h \times (d_h + d_{\text{in}})}\), \(b_r \in \mathbb{R}^{d_h}\)
- \(r_t \in (0, 1)^{d_h}\)
Per-dimension behaviour:
- \(r_t^{(j)} \approx 1\): past visible to candidate
- \(r_t^{(j)} \approx 0\): past ignored by candidate
Update gate
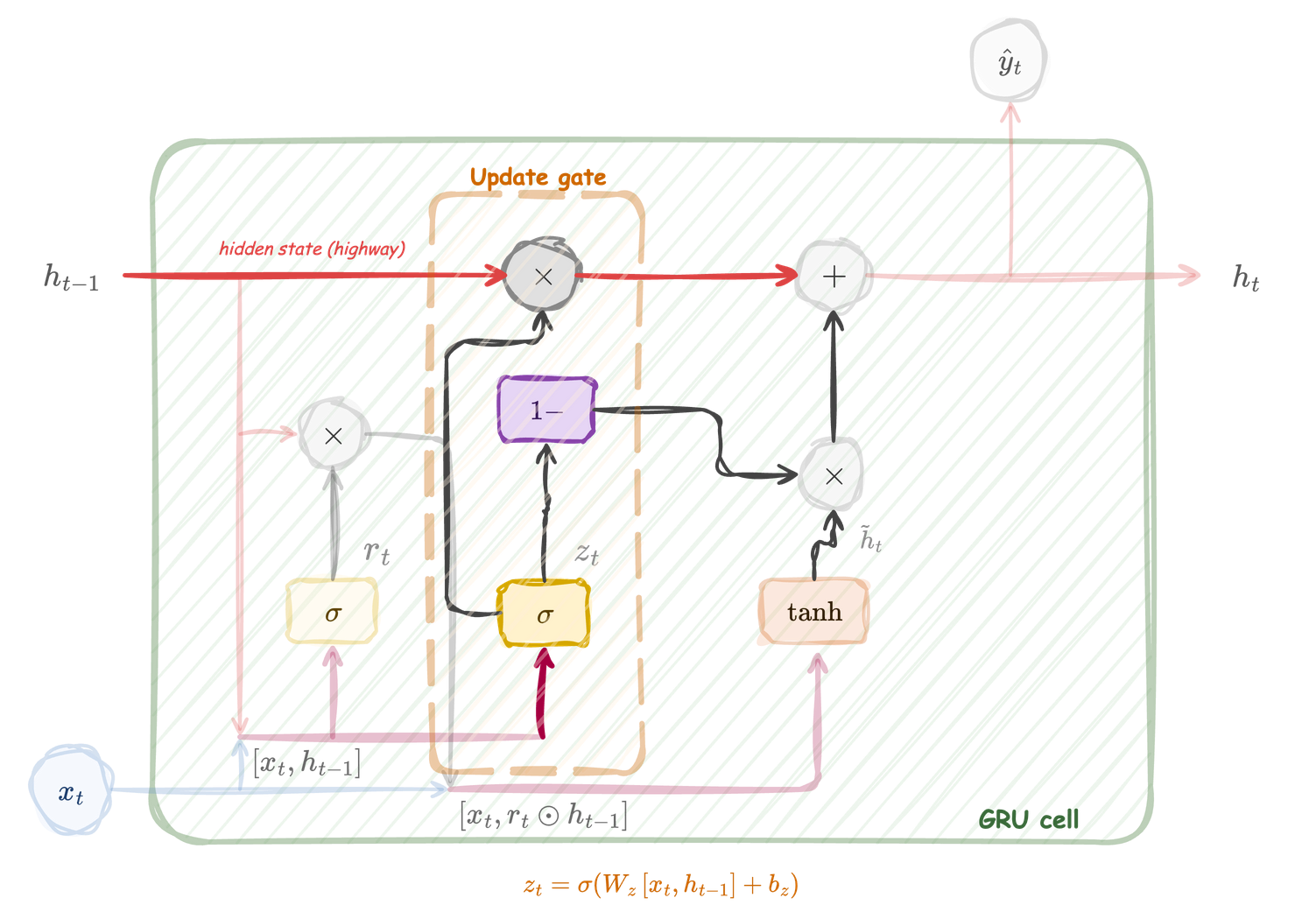
The update gate reads \([h_{t-1}, x_t]\) and produces \(z_t\) through a sigmoid. It controls the interpolation between the old state \(h_{t-1}\) and the new candidate \(\tilde{h}_t\). A value of \(z_t \approx 1\) means “keep the old state”; \(z_t \approx 0\) means “replace with the candidate”.
Update gate
\[ z_t = \sigma \bigl(W_z\, [h_{t-1},\, x_t] + b_z\bigr) \]
Dimensions:
- \(W_z \in \mathbb{R}^{d_h \times (d_h + d_{\text{in}})}\), \(b_z \in \mathbb{R}^{d_h}\)
- \(z_t \in (0, 1)^{d_h}\)
Dual role:
- \(z_t\) controls retention of \(h_{t-1}\)
- \((1 - z_t)\) controls admission of \(\tilde{h}_t\)
- One gate replaces both LSTM forget and input gates

Putting it all together

Complete GRU cell. The reset gate modulates \(h_{t-1}\) for the candidate; the update gate interpolates between old and new states.
Putting it all together
All three equations for one time step:
\[\begin{eqnarray} r_t &=& \sigma\bigl(W_r [h_{t-1}, x_t] + b_r\bigr) \\ z_t &=& \sigma\bigl(W_z [h_{t-1}, x_t] + b_z\bigr) \\ \tilde{h}_t &=& \tanh\bigl(W_h [r_t \odot h_{t-1}, x_t] + b_h\bigr) \\ h_t &=& z_t \odot h_{t-1} + (1 - z_t) \odot \tilde{h}_t \end{eqnarray}\]
Input: \(h_{t-1}, x_t\). Output: \(h_t\).
Worked example: sequence classification with an LSTM
Task: classify protein subcellular localisation from amino-acid sequences.
- Input: one-hot or embedding of amino acids, \(x_t \in \mathbb{R}^{d_{\text{in}}}\)
- Sequence length: variable (\(T\) ranges from tens to thousands)
- Output: one of \(k\) compartments (nucleus, cytoplasm, membrane, …)
Architecture
\[ x_t \;\xrightarrow{\text{embedding}}\; e_t \in \mathbb{R}^{d_e} \;\xrightarrow{\text{LSTM}}\; h_t \in \mathbb{R}^{d_h} \;\xrightarrow{\text{pool + linear}}\; z \in \mathbb{R}^{k} \;\xrightarrow{\text{softmax}}\; \hat{y} \]
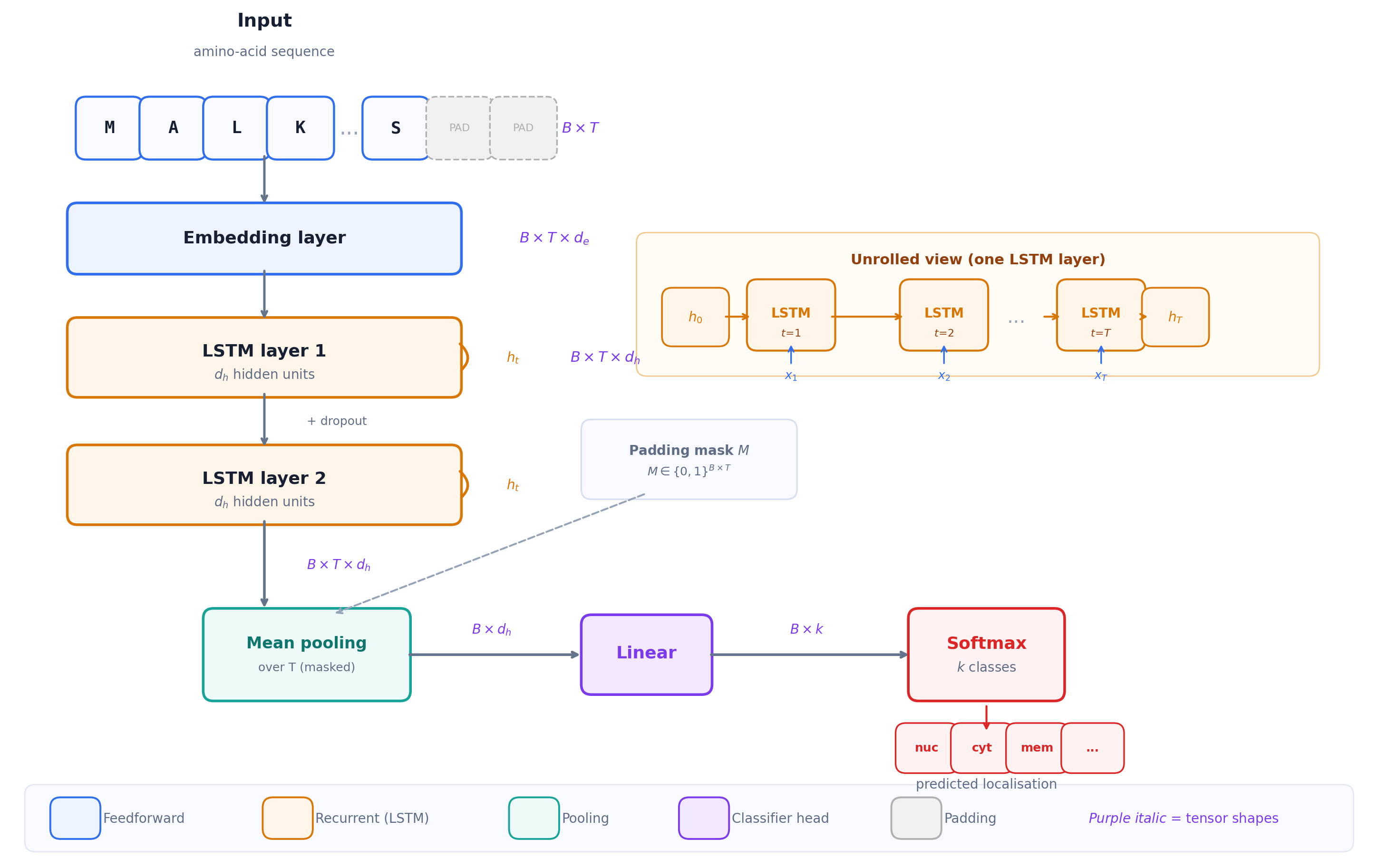
LSTM sequence classifier with embedding, recurrent layers, pooling, and softmax output
Questions

References
Anfinsen, Christian B. 1973. “Principles That Govern the Folding of Protein Chains.” Science 181: 223–30.
Bahdanau, Dzmitry, Kyunghyun Cho, and Yoshua Bengio. 2015. “Neural Machine Translation by Jointly Learning to Align and Translate.” arXiv Preprint arXiv:1409.0473.
Cho, Kyunghyun, Bart van Merriënboer, Caglar Gulcehre, et al. 2014. “Learning Phrase Representations Using RNN Encoder–Decoder for Statistical Machine Translation.” arXiv Preprint arXiv:1406.1078.
Chung, Junyoung, Caglar Gulcehre, Kyunghyun Cho, and Yoshua Bengio. 2014. “Empirical Evaluation of Gated Recurrent Neural Networks on Sequence Modeling.” arXiv Preprint arXiv:1412.3555.
ENCODE Project Consortium. 2012. “An Integrated Encyclopedia of DNA Elements in the Human Genome.” Nature 489: 57–74.
Hochreiter, Sepp, and Jürgen Schmidhuber. 1997. “Long Short-Term Memory.” Neural Computation 9 (8): 1735–80.
Hornik, Kurt, Maxwell Stinchcombe, and Halbert White. 1989. “Multilayer Feedforward Networks Are Universal Approximators.” Neural Networks 2: 359–66.
Jozefowicz, Rafal, Wojciech Zaremba, and Ilya Sutskever. 2015. “An Empirical Exploration of Recurrent Network Architectures.” Proceedings of the 32nd International Conference on Machine Learning (ICML), 2342–50.
Jumper, John et al. 2021. “Highly Accurate Protein Structure Prediction with AlphaFold.” Nature 596: 583–89.
Madani, Ali et al. 2023. “Large Language Models Generate Functional Protein Sequences Across Diverse Families.” Nature Biotechnology 41: 1099–106.
Pascanu, Razvan, Tomas Mikolov, and Yoshua Bengio. 2013. “On the Difficulty of Training Recurrent Neural Networks.” Proceedings of the 30th International Conference on Machine Learning, 1310–18.
Sutskever, Ilya, Oriol Vinyals, and Quoc V. Le. 2014. “Sequence to Sequence Learning with Neural Networks.” Advances in Neural Information Processing Systems 27.
Werbos, Paul J. 1990. “Backpropagation Through Time: What It Does and How to Do It.” Proceedings of the IEEE 78 (10): 1550–60.
